Болезнь Шарко — Мари — Тута
| Болезнь Шарко — Мари — Тута | |
|---|---|
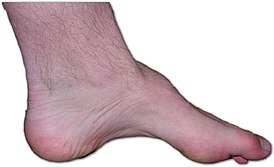 Стопа пациента с характерными симптомами болезни Шарко — Мари — Тута: дистрофия мышц, полая стопа, молоткообразные пальцы. | |
| МКБ-10 | G60.0 |
| МКБ-10-КМ | G60.0 |
| МКБ-9 | 356.1 |
| МКБ-9-КМ | 356.1 |
| OMIM | 118220 |
| DiseasesDB | 5815 и 2343 |
| MedlinePlus | 000727 |
| MeSH | D002607 |
Болезнь Шарко — Мари — Тута (ШМТ), или наследственная моторно-сенсорная нейропатия (НМСН) — наследственная периферическая нейропатия с хроническим прогрессирующим течением. При этой болезни больные страдают от слабости и атрофии мышц дистальных отделов конечностей, деформации стоп и кистей, у них наблюдается снижение сухожильных рефлексов, изменение походки, потеря чувствительности в конечностях. В основе клинических проявлений болезни лежит поражение двигательных и чувствительных периферических нервных волокон. Болезнь Шарко — Мари — Тута диагностируется с приблизительной частотой 1 на 2500 человек. Первое проявление болезни чаще всего происходит в подростковом возрасте или в раннем взрослом возрасте. Тяжесть симптомов сильно различается даже среди членов одной семьи с этим заболеванием. Болезнь Шарко — Мари — Тута нередко ведёт к ограничению трудоспособности и к инвалидизации, при этом большинство пациентов имеют нормальную продолжительность жизни. Болезнь Шарко — Мари — Тута является генетически крайне неоднородным заболеванием, симптомы этой болезни могут быть вызваны мутациями в более чем двух десятках генов, хотя большая часть заболеваний вызвана мутациями в генах PMP22, MPZ, GJB1 и MFN2. Наследование болезни чаще всего аутосомно-доминантное, однако может быть аутосомно-рецессивным и Х-сцепленным.
Заболевание носит имена врачей, впервые его описавших в 1886 году: французских врачей Жана-Мартена Шарко и Пьера Мари, а также англичанина Говарда Тута.
Основные формы болезни Шарко — Мари — Тута
Существуют различные формы болезни Шарко — Мари — Тута. Основные формы имеют обозначения ШМТ1, ШМТ2, ШМТ3, ШМТН4, ШМТ5, ШМТ6, ШМТ-ДП, ШМТ-РП и ШМТХ.
Причиной ШМТ1 является нарушение миелиновой оболочки периферических нервов, эта форма называется миелинопатия и имеет несколько типов со сходными симптомами. Первые признаки болезни появляются, как правило, в подростковом возрасте. Пациенты испытывают мышечную слабость в ногах, у них происходит атрофия мышц дистальных отделов нижних конечностей, где позднее слабеет и утрачивается чувствительность. Скорость проведения импульса по срединному нерву снижена и составляет менее 38 м/с. У пациентов выявляется сегментарная демиелинизация и ремиелинизация. При биопсии нервных волокон выявляется гиперплазия шванновских клеток с формированием характерного морфологического признака «луковичные головки».
- Наиболее распространённый тип ШМТ1А (OMIM#118220) имеет аутосомно-доминантное наследование и вызван дупликацией участка короткого плеча 17-й хромосомы (17p11.2). Этот участок несёт ген PMP22, кодирующий белок PMP22, который является критическим компонентом миелиновой оболочки периферических нервных волокон. В результате дупликации и увеличения дозы гена количество производимого белка PMP22 также увеличивается, что приводит к структурным и функциональным нарушениям миелиновой оболочки.
- ШМТ1B (OMIM#118200) — заболевание с аутосомно-доминантным наследованием, оно вызвано мутацией в гене MPZ, который кодирует белок P0, который является ещё одним важным компонентом миелиновой оболочки. Большинство мутаций, ведущих к развитию патологического фенотипа, являются точечными мутациями. На сегодняшний день ученые выявили более 120 различных точечных мутаций в гене P0.
- Менее распространённые ШМТ1C (OMIM#601098), ШМТ1D (OMIM#607678) и ШМТ1F (OMIM#607734), вызваны мутациями в генах LITAF, EGR2 и NEFL, соответственно.